Introduction¶
Welcome to the user guide describing StochPy: Stochastic modeling in Python. We could start by explaining that StochPy is comprehensive and user-friendly, as we explain in our manuscript, but we decided to start this user guide with a simple, yet illustrative, example:
>>> import stochpy
>>> smod = stochpy.SSA()
>>> smod.DoStochSim()
>>> smod.PlotSpeciesTimeSeries()

‘Nuff said right?
In the remainder of this user guide, we explain e.g. how to:
- Install StochPy: click here,
- Use StochPy in more detail click here,
- Do stochastic simulations with cell growth and division click here.
Installation and Configuration¶
Installation¶
Dependencies¶
StochPy (since version 2.1.0) is available for both Python 2 (2.6+) and Python 3 (3.4+). We recommend using Python 2.7. In addition to Python 2 or 3, the NumPy library is required before installing StochPy.
The following software is optional but recommended:
Matplotlib is useful for the generation of plots and libSBML is necessary to convert models written in SBML format to the PySCeS MDL. The libSBML software requires a XML parser library (libxml2).
On Ubuntu you can use the following code to directly install all required packages on Python 2.6+:
sudo apt-get install python-dev python-numpy
sudo apt-get install python-scipy python-matplotlib python-pip
sudo apt-get install ipython ipython-notebook
sudo apt-get install libxml2 libxml2-dev
sudo apt-get install zlib1g zlib1g-dev
sudo apt-get install bzip2 libbz2-dev
sudo pip install python-libsbml
and for Python 3.4+:
sudo apt-get install python3-dev python3-numpy
sudo apt-get install python3-scipy python3-matplotlib python3-pip
sudo apt-get install ipython3 ipython3-notebook
sudo apt-get install libxml2 libxml2-dev
sudo apt-get install zlib1g zlib1g-dev
sudo apt-get install bzip2 libbz2-dev
sudo pip3 install python-libsbml
Windows¶
Download the windows installer (32-bit) or the source code and use setup.py in a terminal.
Python 2.6.x:
$ python setup.py install
Python 3.4.x:
$ python3 setup.py install
If you download the source code, you can also generate your own windows installer with the following code:
$ python setup.py bdist_wininst
Linux/MAC OS/Cygwin¶
Download the source code and use setup.py in the terminal:
$ cd /.../StochPy-2.3.0
Python 2.7.x:
$ sudo python setup.py install
Python 3.4.x:
$ sudo python3 setup.py install
Configuration¶
The StochPy package contains some example models, such as:
- Immigration-Death model
- Burst model
- Decaying-Dimerizing model
- Prokaryotic auto-regulation model
The files describing these models can be found in the directory /home dir/Stochpy/pscmodels/ after importing stochpy for the first time. Most of these models are written in the PySCeS MDL format. This PySCeS MDL is explained in the PySCeS Model Description Language section. Besides PySCeS MDL, StochPy also accepts models written in SBML.
Using StochPy¶
An interactive Python shell is necessary to start interactive modeling with StochPy. There exist various options, but we recommend using iPython (and iPython notebook for teaching/code sharing). In the command-line you start IPython with the command:
$ ipython
Then, use import stochpy to import the installed version of StochPy:
>>> import stochpy
#######################################################################
# #
# Welcome to the interactive StochPy environment #
# #
#######################################################################
# StochPy: Stochastic modeling in Python #
# http://stochpy.sourceforge.net #
# Copyright(C) T.R Maarleveld, B.G. Olivier, F.J Bruggeman 2010-2015 #
# DOI: 10.1371/journal.pone.0079345 #
# Email: tmd200@users.sourceforge.net #
# VU University, Amsterdam, Netherlands #
# Centrum Wiskunde Informatica, Amsterdam, Netherlands #
# StochPy is distributed under the BSD license. #
#######################################################################
Version 2.3.0
Output Directory: /home/user/Stochpy
Model Directory: /home/user/Stochpy/pscmodels
The message above indicates that the StochPy package is successfully imported and that StochPy is ready-to-use. Use one of the following commands to read StochPy’s docstring:
>>> help(stochpy)
>>> stochpy?
Press q to close the help modus. This functionality is available for every high-level function implemented in StochPy:
>>> smod = stochpy.SSA()
>>> help(smod)
>>> help(smod.DoStochSim)
>>> smod.DoStochSim?
of which DoStochSim() is one of the key high-level functions as it runs a stochastic simulation.
Module 1: Demo¶
Before we explain in more detail how to use StochPy, we want to briefly discuss the demo and utilities modules. The StochPy Demo module currently provides 11 demo’s which illustrate many features of StochPy. In each demo we show the high-level functions that are necessary to obtain the generated results. Information about the used high-level function is shown after the #. Usage is straightforward:
>>> import stochpy
>>> stochpy.Demo()
press any button to continue
Press any button to continue to the first demo:
>>> smod = stochpy.SSA() # start SSA module
>>> smod.DoStochSim(IsTrackPropensities=True)
>>> smod.data_stochsim.simulation_endtime
>>> smod.data_stochsim.simulation_timesteps
>>> smod.PrintWaitingtimesMeans()
Reaction Mean Waiting times
R1 0.109
R2 0.108
>>> smod.PlotSpeciesTimeSeries()
>>> smod.PlotPropensitiesTimeSeries()
>>> smod.PlotWaitingtimesDistributions()
>>> smod.Export2File(analysis="timeseries",datatype="species")
>>> smod.Export2File(analysis="distributions",datatype="species")
>>> smod.Export2File(datatype="propensities")
In the first demo we determine the mean waiting times, export data to text files, and create several plots. You can continue running the remaining demo’s or exit the demo session by pressing CTRL-c.
Module 2: Utilities¶
The StochPy Utilities module contains functions and examples created by users of StochPy:
>>> utils = stochpy.Utils()
>>> utils.DoExample4()

In the future more functions and examples are added to this module.
Module 3: Stochastic Simulation Algorithm¶
The main module of this software package is the stochastic simulation algorithm module. In this module stochastic simulations and their analysis can be done. StochPy currently contains various Stochastic Simulation Algorithms (SSAs):
- Direct Method
- First Reaction Method
- Next Reaction Method (Gibson and Bruck, 1999, J. Phys. Chem.)
- Tau-leap Method (Cao et al. 2006, J. Chem. Phys.), further explained here
- Delayed SSAs, explained here
- Single Molecule Methods, explained here
Run a Stochastic simulation¶
Use the following command to start to the SSA module and to perform your first stochastic simulation with StochPy:
>>> import stochpy
>>> smod = stochpy.SSA()
>>> smod.DoStochSim()
>>> smod.PlotSpeciesTimeSeries()
StochPy uses direct method as default SSA and the immigration-death model as default stochastic model. You can therefore directly use the high-level function DoStochSim() to run your first stochastic simulation. The print statements are suppressed during the simulation. Use quiet=False to enable print statements during the stochastic simulation (see also the Quiet modus section):
>>> smod.DoStochSim(quiet=False)
Info: 1 trajectory is generated
simulation done!
Info: Number of time steps 1000 End time 50.275966611
Info: Simulation time 0.07495
The high-level function DoStochSim() accepts many arguments (e.g. method, trajectories, mode), but we first focus on selecting a stochastic model. Selecting a stochastic model of choice is done with the high-level function Model():
>>> smod.Model("Burstmodel.psc")
>>> smod.Model(model_file="Burstmodel.psc",dir="/home/user/Stochpy/pscmodels")
which accepts besides models described in the PySCeS MDL, also models described in SBML if the appropriate dependencies are installed):
>>> smod.Model("dsmts-001-01.xml",smod.model_dir)
Info: single compartment model: locating "Death" in default compartment
Info: single compartment model: locating "Birth" in default compartment
Writing file: /home/user/Stochpy/pscmodels/dsmts-001-01.xml.psc
>>> smod.model_file
"dsmts-001-01.xml.psc"
>>> smod.model_dir
"/home/user/Stochpy/pscmodels"
After selecting your stochastic model of choice, you can directly use DoStochSim() and specify the method, number of trajectories, simulation mode etc.:
>>> smod.DoStochSim(method="FRM",trajectories=5,mode="time",end=50)
>>> smod.sim_method
stochpy.implementations.FirstReactionMethod.FirstReactionMethod
>>> smod.sim_mode
"time"
Here, we first used the first reaction method to do a stochastic simulation until t=50 (a.u.). Be careful with specifying a certain end time because it’s difficult to predict how long the simulation is going to take. There can be ten time steps in a particular time interval, but this can be just as easily ten million time steps.
Each argument of DoStochSim() is treated as a “local” setting, i.e. they are forgotten if you perform a new stochastic simulation without these arguments:
>>> smod.DoStochSim()
>>> smod.sim_method
stochpy.implementations.DirectMethod.DirectMethod
>>> smod.sim_mode
"steps"
DoStochSim() accepts many more arguments, but we are not going to discuss each of them here in detail. The most used arguments of DoStochSim() are probably: method, mode, end, trajectories, rate_selection, species_selection, IsTrackPropensities, and IsOnlyLastTimepoint. The memory reduction arguments rate_selection, species_selection, and IsOnlyLastTimepoint are discussed here. We refer to the docstring of the high-level function for more information about its arguments:
>>> help(smod.DoStochSim)
Press q to close the docstring.
Alternatively, we can use various high-level functions that allow for specifying “global” simulation settings. These settings are stored and used until you specify them again or load a new model:
>>> smod.Method("Tauleap")
>>> smod.Mode("Time")
>>> smod.sim_method
stochpy.implementations.TauLeaping.OTL
>>> smod.sim_mode
"time"
>>> smod.DoStochSim()
>>> smod.sim_method
stochpy.implementations.TauLeaping.OTL
>>> smod.sim_mode
"time"
While we used high-level function Mode() in the previous example, it is probably easier to directly use one of the following high-level functions:
>>> smod.Endtime(50)
>>> smod.Timesteps(10**5)
We already showed how to select various different stochastic simulation algorithms. The high-level function Method() accepts the following case-insensitive strings: “Direct”, “NextReactionMethod” or “NRM”, “FirstReactionMethod” or “FRM”, “Tauleap”, “DelayedDirect” and “Delayed”, “DelayedNRM” or “DelayedNextReactionMethod”, “SingleMoleculeMethod” or “SMM”, and “FastSingleMoleculeMethod or “fSMM”.
The direct, next reaction method, and first reaction method are exact SSAs that can suffer from extensive running times. If extensive running times are an issue, we recommend using the inexact SSA: the tau-leap method (click here). We also provide exact SSAs with delays (click here) and single molecule methods (here).
Importantly, the high-level function DoStochSim() supports only the simulation of the direct, first reaction, and next reaction, and the tau-leap method. Different high-level functions are necessary for the simulation of e.g. delayed SSAs.
>>> smod.DoStochSim()
>>> smod.DoDelayedStochSim()
>>> smod.DoSingleMoleculeStochSim()
If the wrong method is provided, the high-level function switches to its default method. For example, DoStochSim() switches back to the direct method if e.g. the delayed direct method is provided:
>>> smod.DoStochSim(method="DelayedDirect")
*** WARNING ***: (DelayedDirectMethod) was selected.
Switching to the direct method.
Additionally, StochPy can modify both parameter values and initial species copy numbers with the following high-level functions:
>>> smod.ChangeParameter("Ksyn",5)
>>> smod.ChangeInitialSpeciesCopyNumber("mRNA",100)
Both ChangeParameter() and ChangeInitialSpeciesCopyNumber() change parameter values and species copy numbers in the current StochPy object. Permanent changes can be made by adjusting the model description file (in the directory where the model is stored) and reload the model with the following high-level function:
>>> smod.Reload()
Parsing file: /home/user/Stochpy/pscmodels/ImmigrationDeath.psc
We provide high-level functions that gives a summary of the status of the current settings:
>>> smod.ShowSpecies()
["mRNA"]
>>> smod.ShowOverview()
Current Model: ImmigrationDeath.psc
Number of time steps: 1000
Current Algorithm: <class "stochpy.DirectMethod.DirectMethod">
Number of trajectories: 1
Propensities are not tracked
Here, ShowSpecies() gives a list of all the species in the model and ShowOverview() gives all the current settings. It is also possible to save an interactive session to a Python script:
>>> stochpy.SaveInteractiveSession("interactiveSession1.py")
Reloading the interactive session is done by starting this script in iPython:
$ ipython interactiveSession1.py
Or by running the script inside iPython (make sure you are in the correct working directory):
>>> run interactiveSession1.py
Build-in Analysis Techniques¶
StochPy provides many preprogrammed analysis techniques that can be used to analyze the done stochastic simulations. We start with a discrete time series plot:
>>> smod.Model("ImmigrationDeath.psc")
>>> smod.DoStochSim(method="direct",mode="time",end=50)
>>> smod.PlotSpeciesTimeSeries()
This function accepts many arguments of which species to plot and mark-up details are examples:
>>> smod.PlotSpeciesTimeSeries(xlabel="Time (s)",title="",
linestyle="dashed",linewidth=1,
colors="red",IsLegend=True)
By default, StochPy creates a figure legend. This legend can be (i) turned off with IsLegend =False or (ii) moved to a different location with legend_location. The default legend location is the upper right corner. Both IsLegend and legend_location are arguments of each plotting function. We refer to the docstring of the respective high-level function for more information.
Plots are directly shown if the interactive environment is successfully loaded. Using interactive plotting on the operating systems Windows can cause figures to freeze. If you encounter this issue, please have a look at this section (changing the Matplotlib backend solves typically the problem). It is also possible to turn off the interactive plotting feature:
>>> smod = stochpy.SSA(IsInteractive = False)
With the interactive modes turned off, plotting is possible but the figures are not directly shown. The first command shows the figure (if possible) and the second command saves it to a file:
>>> stochpy.plt.show()
>>> stochpy.plt.savefig("myfigure.pdf")
Before we continue with the remaining preprogrammed analysis techniques, we want to briefly discuss a general problem of stochastic simulations: What is the right number of time steps to get an accurate prediction of e.g. the moments or a distribution? StochPy provides a function, DoCompleteStochSim(), that continues until the first four moments converge within a user-specified error (default = 0.001).
>>> smod.DoCompleteStochSim()
We can use the output of this simulation to get an accurate prediction of the species probability distribution:
>>> smod.PlotSpeciesDistributions()

We recommend using at least one million time steps before analyzing a probability distribution. Another (related) analysis feature offered by StochPy is determining both the mean and standard deviation of each species in the simulated model:
>>> smod.PrintSpeciesMeans()
Species Mean
mRNA 50.086
>>> smod.PrintSpeciesStandardDeviations()
Species Standard Deviation
mRNA 7.079
Calculating the mean and standard deviation of each species in a stochastic simulation is not as straightforward as it may sound. The time between two events is not constant, thus it is necessary to track the time spent in each state for each species. These species statistics can also be accessed via the data_stochsim model object as Python dictionaries:
>>> smod.data_stochsim.species_means
{"mRNA": 50.086035161133793}
>>> smod.data_stochsim.species_standard_deviations
{"mRNA": 7.0793421388907829}
Also, StochPy allows the calculation and analysis of event waiting times. Event waiting times are the times between two subsequent firings of a particular reaction. StochPy can offer this unique feature, because StochPy stores all reaction events.
>>> smod.GetWaitingtimes()
>>> smod.PlotWaitingtimesDistributions()
>>> stochpy.plt.xlim(xmin=10**-5)
>>> smod.PrintWaitingtimesMeans()
Reaction Mean
R1 0.100
R2 0.100
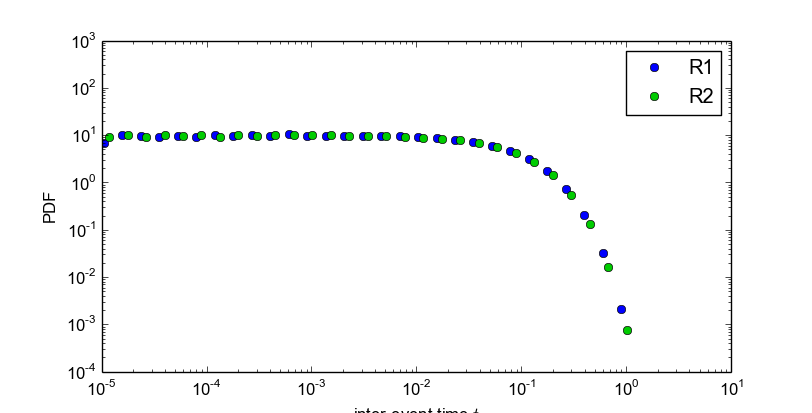
Furthermore, StochPy can determine propensities at each time point during the simulation. Propensities data is not stored by default. The boolean argument IsTrackPropensities of the high-level function DoStochSim() must be set to True:
>>> smod.DoStochSim(IsTrackPropensities=True)
>>> smod.PlotPropensitiesTimeSeries()
>>> smod.PlotPropensitiesDistributions()
This means that we can generate two types of time series data: species and propensities.
The Gillespie algorithms generate data at irregular time points, but StochPy also offers an approach which returns data on a fixed regular time grid. A grid of which the user can specify the resolution (n_samples). The formation of such a regular grid allows for the analysis of autocorrelation (and autocovariance) of species and/or propensities within StochPy. Autocorrelation is the cross-correlation, a measure of similarity of a signal with itself. For different time separations (lags), this similarity can be determined which results a value between 1 and -1. Here, 1 corresponds to positive correlation and -1 to negative correlation. Typically, the first 50 lags are of interest. Here, we show the first 20 lags:
>>> smod = stochpy.SSA()
>>> smod.Model("ImmigrationDeath.psc")
>>> smod.DoStochSim(end=1000,mode="time",IsTrackPropensities=True)
>>> smod.GetSpeciesAutocorrelations(n_samples=1001)
>>> smod.PlotSpeciesAutocorrelations(nlags=20)
>>> smod.GetPropensitiesAutocorrelations()
>>> smod.PlotPropensitiesAutocorrelations(nlags=20)
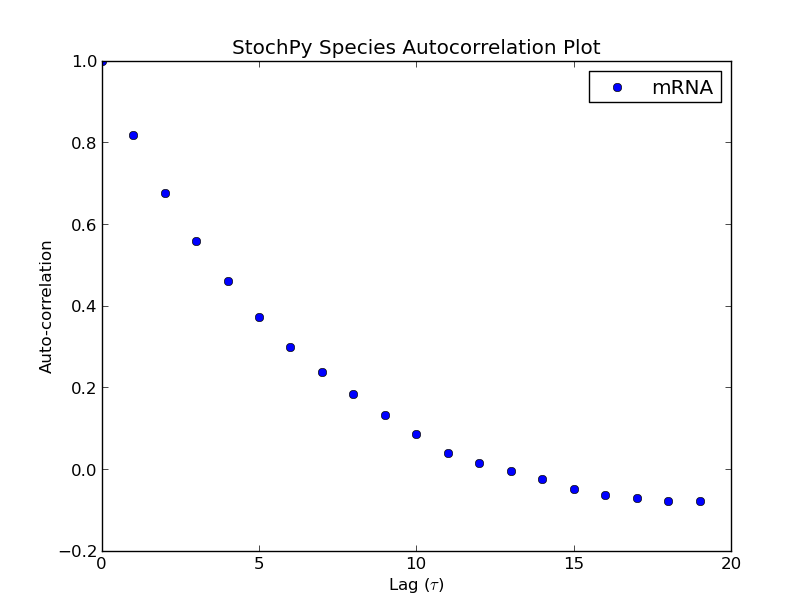
Additionally, we can use the regular grid for analyzing multiple generated trajectories. Via StochPy, we can generate e.g. average time series plots for both species and propensities. Here, we discuss two different plotting functionalities for analyzing probability distributions of species averaged over multiple trajectories. The argument nstd can be used to set the number of standard deviations of the error bars:
>>> smod.DoStochSim(trajectories=10,end=10**5)
>>> smod.GetRegularGrid(n_samples=51)
>>> smod.PlotAverageSpeciesDistributions()
>>> smod.PlotAverageSpeciesDistributionsConfidenceIntervals(nstd=2)
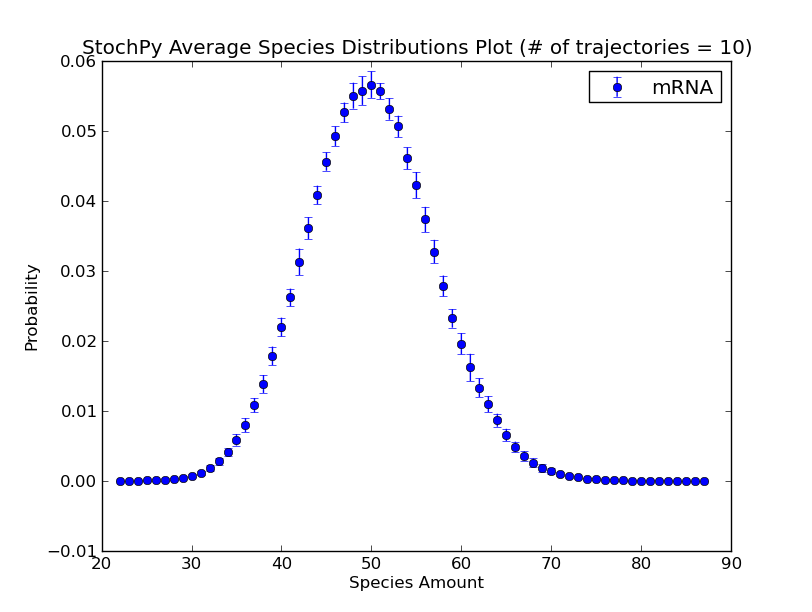

We already explained that we do not have to store the molecule copy numbers of every species. As a consequence, we can only plot data for “stored” species:
>>> smod.PlotSpeciesTimeSeries(species2plot = ["S1","S2"])
>>> smod.PlotPropensitiesTimeSeries(rates2plot = "R1")
>>> smod.PlotWaitingtimesDistributions(rates2plot = "R2")
>>> smod.PlotSpeciesDistributions("S4")
AssertionError: Species S4 is not in the model
StochPy also offers stochpy.plt (a renamed version of matplotlib.pyplot) to manipulate generated plots or to generate your own plots:
>>> smod.PlotSpeciesTimeSeries(marker = "v")
>>> stochpy.plt.title("Your own title")
>>> stochpy.plt.xlabel("Time (s)")
>>> stochpy.plt.xlim([0,10**3])
>>> stochpy.plt.savefig("stochpy_plot.pdf")
Besides plotting features StochPy allows printing and exporting of generated data sets. Printing data is only useful for small datasets, so we prefer using Export2File() which allows data storage in a text file for e.g. external manipulation:
>>> smod.Export2File()
>>> smod.Export2File(analysis="timeseries",datatype="species")
>>> smod.Export2File(analysis="timeseries",datatype="propensities")
>>> smod.Export2File(analysis="distributions",datatype="species")
>>> smod.Export2File(analysis="distributions",datatype="waitingtimes")
>>> smod.Export2File(analysis="distributions",datatype="autocorrelations")
>>> smod.Export2File(analysis="timeseries",datatype="species",IsAverage=True)
StochPy also supports the import of previously exported time series data:
>>> smod.Import2StochPy("myfile","myfiledir",delimiter="\t")
Finally, one can access and manipulate data objects that store the simulation data to perform your own type of analysis in StochPy. See Using Stochpy as a Library.
Inexact Stochastic Algorithms¶
Here, we use the “decaying-dimerizing” model to illustrate the potential advantage of inexact SSA solvers. While exact solvers always simulate one reaction per time step, inexact solvers such as the tau-leap method try to simulate more than one reaction per time step. They do this by asking the following question: How often does each reaction fire in the next specified time interval tau. The larger the value of this tau the more reactions can fire within this time period, but this tau value must be small enough to make sure that no propensity function suffers from a significant change.
A significant speed advantage can be obtained for models where molecules are present in large quantities. An example of such a system is the decaying-dimerizing model, where S1(t=0)= 100000. We simulate this model with first the tau-leap and secondly the direct method of which the results are shown below. Information about exact firing times (and therefore also event waiting times) cannot be obtained with this tau-leap method:
>>> smod.Model("DecayingDimerizing.psc")
>>> smod.DoStochSim(method="Tauleap",end=50,mode="time") # sim. time 0.51 s
>>> smod.PlotSpeciesTimeSeries()
>>> smod.DoStochSim(method="Direct",end=50,mode="time") # sim. time 27.9 s
>>> smod.PlotSpeciesTimeSeries()
>>> stochpy.plt.xscale("log")

Tau-leap methods exploit a error-control parameter, epsilon, that bounds the expected change in the propensity functions. This epsilon value ranges between 0-1 where smaller values result in more accurate simulations (but also a smaller tau size and longer simulations). We set this value, by default, to 0.03 (as is also done by Cao et al. 2006). For the decaying-dimerizing model, the tau-leap method is about 50 times faster than the direct method. StochPy allows users to manipulate this setting with the argument epsilon, as we show next.
We now set epsilon to 0.05:
>>> smod.DoStochSim(method="Tauleap",end=50,mode="time",epsilon=0.05)
The tau-leap method is now about 100 times faster than the direct method. Too large values of epsilon give incorrect simulation results. We therefore recommend verifying the outcome of the tau-leap method against that of an exact solver. A more strict value can be necessary if you want an accurate prediction of species quantities at different time points. An example is the dsmts-001-05 model of the test suite from Evans et al. 2008. We demanded an accurate prediction at t=0,1, ... 50 for which an epsilon value of 0.01 was required.
* Warning *
The tau-leap method automatically reduces the epsilon value for second and third-order reactions. StochPy, therefore, tries to tries to determine the order of each reaction, the “highest order of reaction” (HOR) for each species and its contribution. This works properly for mass-action kinetics, but is likely to fail for non mass-action kinetics. Use the following commands to access the automatically generated results of StochPy:
>>> smod.Model("DecayingDimerizing.psc")
>>> smod.Method("Tauleap")
>>> smod.SSA.parse.reaction_orders # for each reaction
[1,2,1,1]
>>> smod.SSA.parse.species_HORs # for each species
[2,1,0]
>>> smod.SSA.parse.species_max_influence # for each species
[2,1,0]
These results are correct (the model is also described by mass-action kinetics).
- All reactions are first order, except the second reaction. This is a dimerization reaction of species S1.
- The highest order of reaction (HOR) of S1 is thus 2. Species S2 acts as reactant in a first-order reaction (HOR, S2 = 1) and S3 is not a reactant (HOR, S3 = 0).
- species_max_influence gives the highest contribution of a species to its HOR. The second reaction is, as said, a dimerization reaction of species S1, so the contribution of S1 for this reaction is also 2. In a similar manner, we can conclude that the highest contribution of species S2 to its HOR is 1 (it’s a first-order reaction).
StochPy allows users to modify the three lists shown above directly, but one can also provide them as arguments to the DoStochSim() function:
>>> smod.DoStochSim(end=50,mode="time",method="Tauleap",
reaction_orders=[1,2,1,1],
species_HORs=[2,1,0],
species_max_influence=[2,1,0])
Finally, StochPy”s implementation of the tau-leap method defines “critical reactions”, i.e. reactions that are within a few firings of exhausting (one of) its reactants. These critical reactions are used to reduce the probability for a negative population by allowing these reactions to fire only once per time step. The automated determination of critical reactions works fine for most rate equations, but could go wrong for some very specific rate equations. StochPy allows specification of reactions that can fire no more than once during a single time step for the entire simulation. While it is not necessary for the decaying-dimerizing model, we now set R4 as a critical reaction for the entire simulation:
>>> smod.DoStochSim(end=50,mode="time",method="Tauleap",
critical_reactions=["R4"])
Because it is not necessary for this model, the simulation time increases notably (only five times faster than the direct method).
Delayed Stochastic Algorithms¶
Delayed stochastic algorithm extent the Gillespie stochastic simulation algorithm (SSA) to a system with delays. A delayed reaction consists of an exponential waiting time as initiation step with a subsequent delay time. We implemented both the Delayed Direct Method (Cai 2007, J. Chem. Phys) and the Delayed Next Reaction Method (Anderson 2007, J. Chem. Phys).
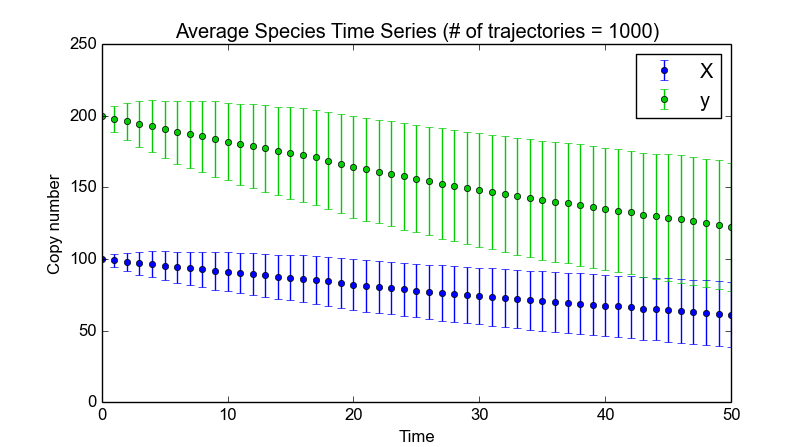
To illustrate the difference between “normal” and “delayed” SSAs, we start by running a normal stochastic simulation with a simple model where X molecules are converted to Y molecules:
>>> smod.Model("Isomerization.psc")
>>> smod.DoStochSim(mode="time",end=10,trajectories=1000)
>>> smod.GetRegularGrid(n_samples=50)
>>> smod.PlotAverageSpeciesTimeSeries()

Next, we set a fixed delay of five seconds on reaction R1. This means that after R1 fires, a X molecule is consumed and it takes exactly five seconds before a Y molecule is produced:
>>> smod.SetDelayParameters({"R1":("fixed",5)})
>>> smod.DoDelayedStochSim(mode="time",end=10,trajectories=1000)
>>> smod.GetRegularGrid(n_samples=50)
>>> smod.PlotAverageSpeciesTimeSeries()

There exist two types of delayed reactions: consuming and nonconsuming delayed reactions. For consuming delayed reactions, the reactants are consumed at initiation and for nonconsuming delayed reactions, the reactants are consumed at completion. In both cases, the products are updated at completion. By default, delayed reactions are “consuming delayed reactions”. Nonconsuming delayed reactions have to be specified specifically with the argument nonconsuming_reactions. In this example, after reaction R1 fires, it takes exactly five seconds before a particular X is degraded and a particular Y is produced:
>>> smod.SetDelayParameters({"R1":("fixed",5)},nonconsuming_reactions=["R1"])
>>> smod.DoDelayedStochSim(mode="time",end=10,trajectories=1000)
>>> smod.GetRegularGrid(n_samples=50)
>>> smod.PlotAverageSpeciesTimeSeries()
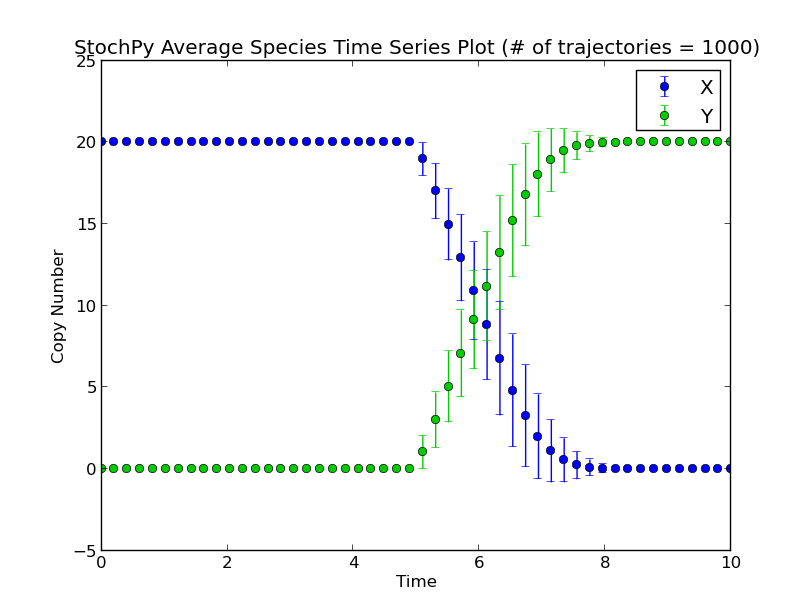
Performing a delayed stochastic simulation is not allowed without setting delayed parameters:
>>> smod.Model("ImmigrationDeath.psc")
>>> smod.DoDelayedStochSim()
AttributeError: No delay parameters have been set.
Please first use the function .SetDelayParameters().
An empty set of delayed parameters is sufficient, but realize that this is similar to doing a non-delayed stochastic simulation:
>>> smod.SetDelayParameters({})
>>> smod.DoDelayedStochSim()
Next, we use a small model of gene transcription to illustrate the difference between consuming and nonconsuming delayed reactions. RNA polymerase can bind to the promoter region of a gene, which starts the transcription process. When the RNA polymerase leaves the promoter region and elongation phase of the transcription process starts, a next RNA polymerase can bind to the gene. This means that, if we assume that the number of RNA polymerases is relatively large, gene transcription can be viewed as a nonconsuming reaction.
Modeling this process as a nonconsuming reactions gives completely different results as is shown here with two simple simulations. First, we simulate this process as a consuming reaction. The available RNA polymerase pool depletes quickly which reduces the formation rate of mRNA chains:
>>> smod.Model("Polymerase.psc")
>>> smod.Endtime(200)
>>> smod.SetDelayParameters({"R1":("fixed",5)})
>>> smod.DoDelayedStochSim()
>>> smod.PlotSpeciesTimeSeries()
>>> stochpy.plt.ylim(0,70)

And second, we model this process as a nonconsuming reaction. Now, the available RNA polymerase stays constant, and therefore, significantly more mRNA chains are produced.
>>> smod.SetDelayParameters({"R1":("fixed",5)},nonconsuming_reactions="R1")
>>> smod.DoDelayedStochSim()
>>> smod.PlotSpeciesTimeSeries()
>>> stochpy.plt.ylim(0,70)

Single Molecule Method¶
In stochastic simulations, reaction times are exponential random variables with mean and standard deviation 1/a0; a0 is the sum of all propensities. For certain processes, reaction times can be distributed differently (think of e.g. lumped reactions). One way to circumvent this is to exploit delays. We developed a method called the Single Molecule Method (SMM) that approaches reactions differently. Here, both the exponential step and a potential delay are replaced by a reaction time for the whole reaction. More importantly, the reaction time does not have to be drawn from an exponential distribution, whereas can be drawn from any distribution.
The propensity is typically a rate parameter multiplied with one or more species. For example, for the first-order reaction X -> Y, the propensity function is k1*X. Hence, the mean reaction time decreases with increasing X and vice versa. The SMM treats the copy number differently: Putative reaction times are determined for each set of substrate molecules individually. This means that, for our example model X -> Y, we determine a putative reaction time for each X molecule. These putative reactions times can be fixed or drawn from a distribution of choice. Of course, negative values are not allowed so be careful with e.g. normal distributions. If a molecule acts as a reactant in multiple reactions, it has a putative reaction time for each of these reactions. The shortest putative reaction time “wins” and the molecule reacts in this reaction channel. This is similar to the Next Reaction Method developed by Gibson and Bruck, with the difference that we look at single molecule rather than single reactions.
The SMM can be rather slow, especially for models with many second-order reactions with high reactant copy numbers. For second-order reactions, every combination of molecules should be considered. We use a simple example to illustrate this. Consider the reaction A + B > C, with ten molecules of A and hundred molecules of B. There are in total thousand different reactions possible, i.e. each molecule of A can react with hundred different molecules of species B. For each of these thousand reactions, a putative reaction time is generated. After selecting the reaction with the shortest putative reaction time, many modifications have to be made to the system. For instance, all reactions that belong to the selected A and B molecules have to be deleted. Imagine what happens if multiple reactions can use these compounds as reactants.
Often, the majority of reactions in a model are regular exponential reactions. We, therefore developed the fast Single Molecule Method (fSMM) to speed-up the SMM simulation time. This method combines a regular Next Reaction Method and the SMM. The main advantage is that only reactions which are given a particular putative reaction time distribution, the single molecule reactions, are treated as in the SMM. The other reactions have a exponential putative time distribution and are simulated as in a normal Next Reaction Method. Hence, the number of putative reaction times stored are significantly decreased, resulting in a faster search for the shortest putative reaction time.
Before we use a simple example to illustrate the single molecule method, we first discuss a few important points:
- Do not use rate equations different than mass-action kinetics if you use the SMM method (e.g. dsmts-003-05 fails). The fSMM does support rate equations different than mass-action kinetics.
- Do not use third-order rate equations in for both the SMM and the fSMM.
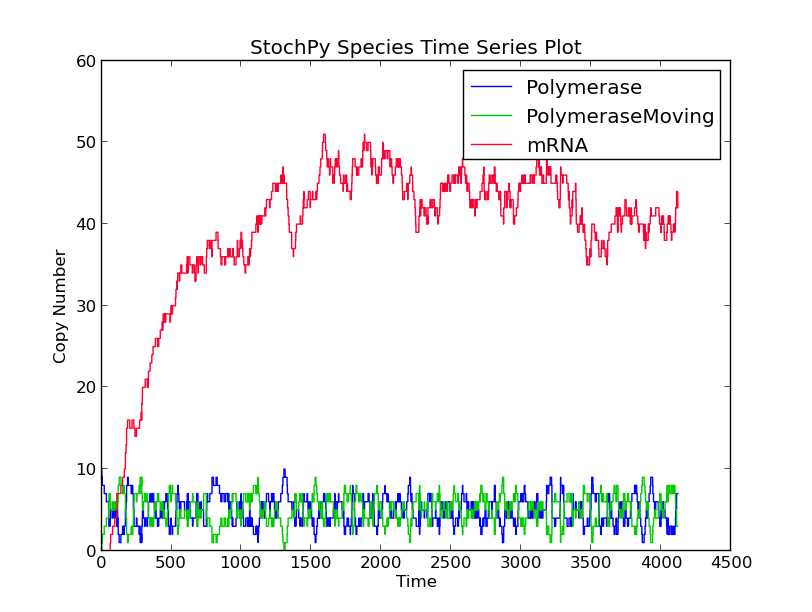
In this first example model, we explicitly model a intermediate form of the polymerase, polymerase moving. After exactly 60 seconds, the moving polymerase produces mRNA and is free again.
>>> smod.Model("TranscriptionIntermediate.psc")
>>> smod.SetPutativeReactionTimes({"R2":("fixed",60)})
>>> smod.DoSingleMoleculeStochSim(method = "fSMM")
>>> smod.PlotSpeciesTimeSeries()
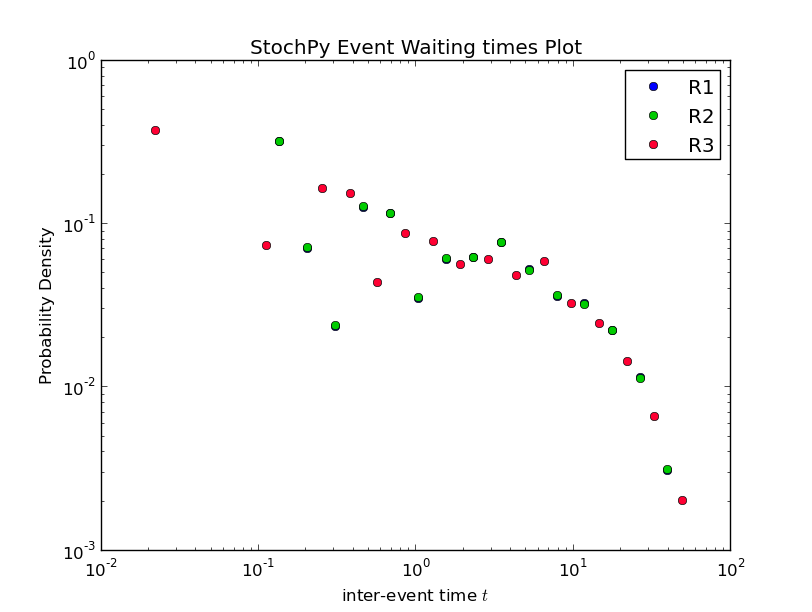
>>> smod.PlotWaitingtimesDistributions()
Interestingly, the waiting times between subsequent firings of R1 and R2 match; R2 fires exactly 60 time units after R1 fires. In a delayed-SSA, the intermediate form is not modeled explicitly.
Examples¶
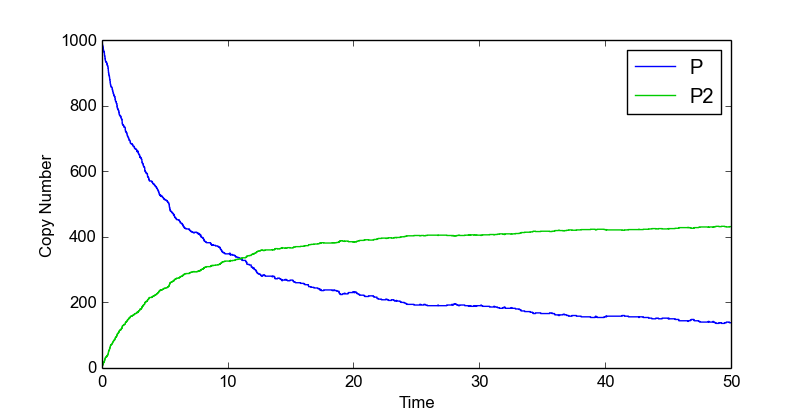
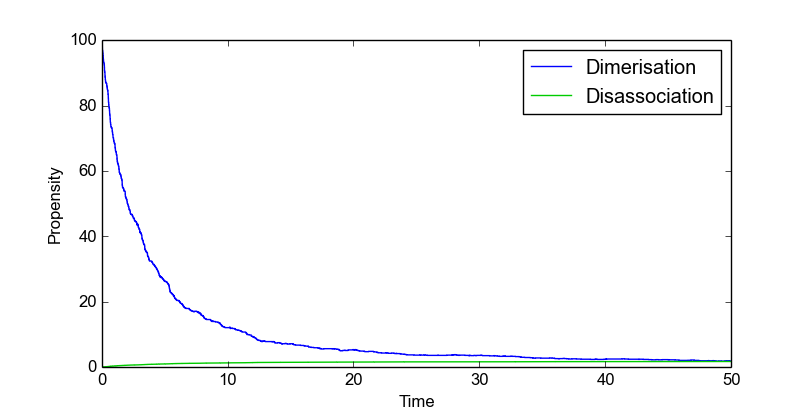
Stochastic simulations are done with a dimerization model to illustrate how you can use the stochastic simulation algorithm module. This model contains two species, denoted by P and P2:
>>> import stochpy
>>> smod = stochpy.SSA()
>>> smod.Model("dsmts-003-02.xml.psc")
>>> smod.DoStochSim(end = 50,mode = "time",IsTrackPropensities=True)
>>> smod.PlotSpeciesTimeSeries()
>>> smod.PlotPropensitiesTimeSeries()
Doing a simulation for more time steps for allows the calculation of more accurate species distributions and event waiting time distributions:
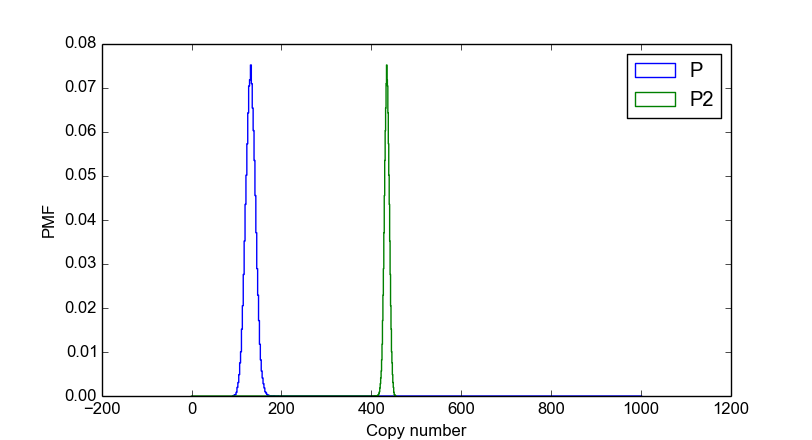
>>> smod.DoStochSim(end=5000,mode="time",IsTrackPropensities=True)
>>> smod.PlotSpeciesDistributions(species2plot="P",bin_size=2,colors="blue")
>>> smod.plot.plotnum-=1 # reset figure number
>>> smod.PlotSpeciesDistributions(species2plot="P2",bin_size=1,colors="green")
>>> stochpy.plt.legend(['P','P2'])
>>> smod.PlotWaitingtimesDistributions()

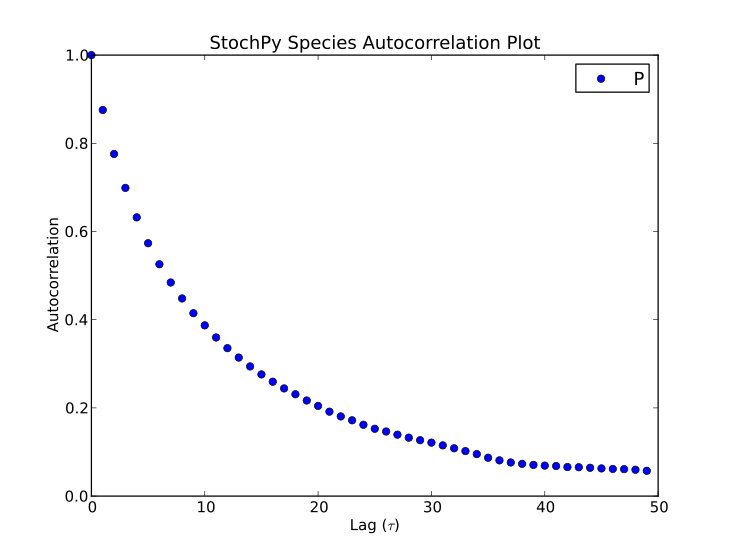
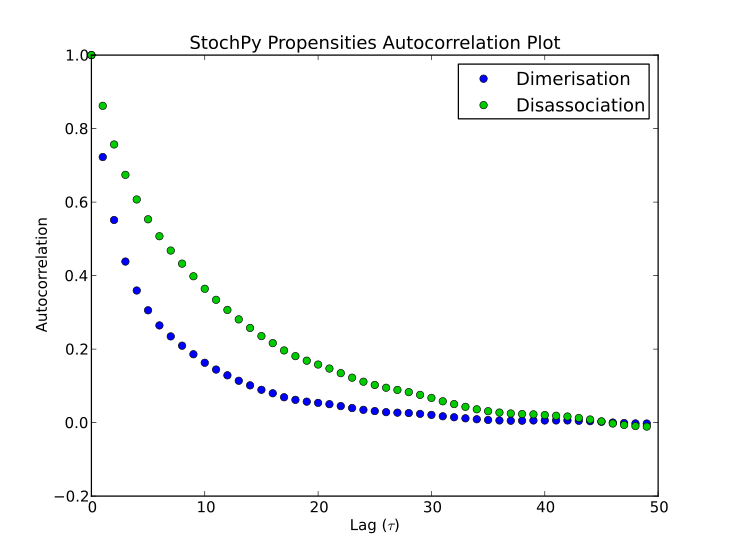
Finally, autocorrelations for both species and propensities can be determined:
>>> smod.GetSpeciesAutocorrelations()
>>> smod.GetPropensitiesAutocorrelations()
>>> smod.PlotSpeciesAutocorrelations(species2plot="P",nlags=50)
>>> smod.PlotPropensitiesAutocorrelations(nlags=50)
Reducing Memory Usage¶
StochPy returns the “raw” stochastic simulation output, i.e. for each firing time we store the copy number of each species and if desired also the propensity of each reaction. This can result in memory issues for models with many species (and reactions). For this reason, StochPy provides several ways to reduce the amount of stored data. We provide two arguments: species_selection and rate_selection which can be used to specify for which species and rates we store its copy number and propensity at each firing time. By default, we store this information for each species and for none of the rates. Adding IsTrackPropensities =True as argument results in storing this information for every rate. Here, we provide an example of a model that consists of 50 species and 98 reactions. We store two species and the propensities of two reactions.
>>> smod.Trajectories(1)
>>> smod.Model("chain50.psc")
>>> smod.DoStochSim(species_selection=["S1","S5"],rate_selection=["R1","R15"])
>>> smod.data_stochsim.species_labels
["S1", "S5"]
>>> smod.data_stochsim.species
array([[ 1., 0.],
[ 0., 0.],
[ 0., 0.],
...,
[ 0., 0.],
[ 0., 0.],
[ 0., 0.]])
>>> smod.data_stochsim.propensities
array([[ 1., 0.],
[ 0., 0.],
[ 0., 0.],
...,
[ 0., 0.],
[ 0., 0.],
[ 0., 0.]])
>>> smod.data_stochsim.propensities_labels
["R1", "R15"]
In addition, the boolean IsOnlyLastTimepoint can be used to store only the last time point of the simulation. When IsOnlyLastTimepoint =True, many functionalities such as plotting and determining statistics are disabled.
>>> smod.Model("ImmigrationDeath.psc")
>>> smod.DoStochSim(end=50,mode="time",IsOnlyLastTimepoint = True)
>>> smod.data_stochsim.species # molecule copy number at (t=50)
array([[55.]])
>>> smod.data_stochsim.time # stored time points (t=50)
array([[50.]])
Quiet Modus¶
The StochPy high-level functionalities can print information which can be useful for (inexperienced) StochPy users.
>>> smod = stochpy.SSA(IsQuiet=False)
Info: Direct method is selected to perform
Parsing file: /home/timo/Stochpy/pscmodels/ImmigrationDeath.psc
Info: No reagents have been fixed
>>> smod.DoStochSim()
Info: 1 trajectory is generated
simulation done!
Info: Number of time steps 1000 End time 510.780391803
Info: Simulation time 0.08498
StochPy 2.3 provides a quiet modus (default = True). In the quiet modus, only essential information is printed:
>>> smod = stochpy.SSA()
>>> smod.SetQuiet()
>>> smod.DoStochSim()
>>> smod = stochpy.SSA(IsQuiet=True)
>>> smod.DoStochSim()
or alternatively:
>>> smod = stochpy.SSA()
>>> smod.DoStochSim(quiet=True)
This functionality also works in different StochPy modules.
Experiencing plotting problems¶
StochPy uses Matplotlib for plotting. If Windows is your operating system, interactive plotting with Matplotlib can result in figure freezing. If you encounter figure freezing, we suggest to try out different back-ends (http://matplotlib.org/faq/usage_faq.html#what-is-a-backend). TkAgg is probably the best for interactive plotting, but is not available on every operating system and/or python environment:
>>> stochpy.plt.switch_backend("TkAgg")
Different non-interactive back-ends are available (e.g. the “Agg”-backend):
>>> stochpy.plt.switch_backend("Agg")
>>> smod.DoStochSim()
>>> smod.PlotSpeciesTimeSeries() # no plot is shown
>>> stochpy.plt.savefig(stochpy.os.path.join(stochpy.output_dir,"test1.png")
If you switch to a non-interactive back-end, make sure to initialize the SSA object with IsInteractive= False:
>>> smod = stochpy.SSA(IsInteractive=False)
>>> smod.DoStochSim()
>>> smod.PlotSpeciesTimeSeries()
>>> stochpy.plt.savefig(stochpy.os.path.join(stochpy.output_dir,"test1.png")
>>> cmod = stochpy.CellDivision(IsInteractive=False)
Plotting in Windows:
- Canopy command prompt: switch to TkAgg
- Canopy interactive data-analysis environment: works nicely with the default Qt4Agg
- Spyder: works nicely with the default Qt4Agg
- Anaconda command prompt: interactive plotting does not work. Use Agg and/or IsInteractive =False.
Combining StochPy with Python Scientific Libraries¶
StochPy is written in Python, so users of StochPy can take advantage of e.g. the scientific libraries that are available in Python. We illustrate this here with the immigration-death model. This immigration-death model contains one species, mRNA. mRNA is synthesized with a constant rate and degraded with a first-order reaction. The immigration-death model is one of the simplest examples of a Poisson process: A stochastic process where events occur continuously and independently of one another.
One of the characteristics of a Poisson process is that the mean and variance are identical. In this example, we use a synthesis rate of 10 and a degradation rate of 0.2, thus both the mean and variance are 50. Here, we first draw N samples from a Poisson distribution with mean = 50. Secondly, we perform a stochastic simulation for N time steps. The initial mRNA copy number is 50. After the simulation, we create a probability density function of the done stochastic simulation. Finally, we create a histogram of the randomly generated Poisson data and add this to the existing plot of the mRNA probability density function. If N is large enough (we use N = 2.5 million), both distributions should be almost identical.
>>> import numpy as np
>>> lambda_ = 50
>>> N = 2500000
>>> data = np.random.poisson(lambda_,N)
>>> smod.ChangeParameter("Ksyn",10)
>>> smod.ChangeParameter("Kdeg",0.2)
>>> smod.Model("ImmigrationDeath.psc")
>>> smod.DoStochSim(end=N,mode="steps")
>>> smod.PlotSpeciesDistributions(linestyle= "solid")
>>> n, bins, patches = stochpy.plt.hist(data, max(data)-min(data),
normed=1, facecolor="green",align="left")
>>> smod.PrintSpeciesMeans()
mRNA 49.912
>>> smod.PrintSpeciesStandardDeviations()
Species Standard Deviation
mRNA 7.073

Using StochPy as a Library¶
It is straightforward to use StochPy as a library in your project. A data object, data_stochsim, is available for those that want to use StochPy as a library or for those that want to do their own analysis. Species, distributions, propensities, simulation time, and waiting times are stored in NumPy arrays and lists. Labels are stored for each of these data types in separate lists. Furthermore, species, propensities, and waiting time statistics are stored in dictionaries and information about the stochastic simulation such as the number of time steps, the simulation end time, and the trajectory is stored. Data is, of course, only available if it is already calculated.
Returning explicit output can result in large data sets. Hence, if multiple time trajectories are generated the data object of each generated trajectory is dumped in a different file on the hard drive by using pickle, an algorithm for serializing and de-serializing a Python object structure. Once necessary for analysis, these data objects are automatically reloaded into StochPy. To improve performance propensities data is not stored by default. Time series visualization can be time-consuming if large data sets are generated. To improve the visualization performance of time series data, by default, not more than 10000 time series points are shown (but you are free to modify this). The high-level function GetTrajectoryData() can be used to access the simulation data of a specific trajectory. By default the latest generated trajectory is not written to disk space, thus accessible without using the high-level function GetTrajectoryData():
>>> import stochpy
>>> smod = stochpy.SSA()
>>> smod.DoStochSim(trajectories = 10,mode = "steps",end = 1000)
>>> smod.data_stochsim
<stochpy.PyscesMiniModel.IntegrationStochasticDataObj object at 0x32cd750>
>>> smod.data_stochsim.simulation_trajectory
10
>>> smod.data_stochsim.time # time array (not shown)
>>> smod.data_stochsim.species # species array (not shown)
>>> smod.data_stochsim.species_labels
>>> smod.data_stochsim.getSpecies() # time + species array (not shown)
>>> smod.GetSpeciesMeans() # for each species
>>> smod.data_stochsim.species_means
>>> smod.data_stochsim.species_distributions # for each species (not shown)
>>> smod.GetWaitingtimes()
>>> smod.data_stochsim.waiting_times # for each reaction (not shown)
>>> smod.GetTrajectoryData(5)
>>> smod.data_stochsim.simulation_trajectory
5
>>> smod.data_stochsim.getDataInTimeInterval(10,5)
Searching (5:10:15)
Out[10]:
array([[ 5.00059882, 224. ],
[ 5.01493943, 223. ],
[ 5.0151249 , 224. ],
...,
[ 14.95863992, 221. ],
[ 14.96716153, 220. ],
[ 14.99282164, 219. ]])
A second data object (data_stochsim_grid) is available that stores data of multiple trajectories:
>>> smod.GetRegularGrid()
>>> smod.data_stochsim_grid.time # time array (not shown)
>>> smod.data_stochsim_grid.species_means # at every t (not shown)
>>> smod.data_stochsim_grid.species_standard_deviations # STDs (not shown)
Getting fixed-interval Output¶
StochPy’s solvers return the raw stochastic simulation output which makes it slower than simulators that return fixed-interval output. Cain and StochKit are examples of stochastic simulators that return fixed-interval output. Returning fixed-interval output results in losing all information about the time between events. Therefore, it is not possible to get information about, for instance, event waiting times.
The number of fixed-intervals, chosen by the user, determines the accuracy of the simulation results. As an example, creating accurate probability distributions of molecule copy numbers requires a number of fixed-intervals similar to the number of time steps in the simulation. Many simulations are required to determine the right number of fixed-interval to obtain accurate probability distributions. On the other hand, fixed-interval output is useful to get an indication of the behavior of your model or to quickly obtain an accurate estimate of the species means and standard deviations.
Because of the flexible design of StochPy, we decided to offer users of StochPy interfaces to both Cain and StochKit solvers (successfully tested with StochKit version 2.0.11). Users can benefit from the speed advantage of the Cain and StochKit solvers in the interactive modeling environment of StochPy. Subsequently, analysis can be done within StochPy. These solvers give wrong output if net stoichiometric coefficients are used in the model description.
To use Cain and StochKit inside of StochPy, one needs to download and install the interfaces InterfaceCain and InterfaceStochKit, respectively. StochKit solvers cannot be used without an installation of StochKit. In addition, usage of StochKit in StochPy requires modification of “InterfaceStochKit.ini” (before installation or after installation in the installation directory of StochPy). After installation of these interfaces, one can use the high-levels functions DoCainStochSim() and DoStochKitStochSim() to use the faster fixed-interval solvers of Cain and StochKit. DoCainStochSim() accepts the following arguments:
>>> smod.DoCainStochSim(endtime=100,frames=10000,trajectories=False,
solver="HomogeneousDirect2DSearch",
IsTrackPropensities=False)
The interface to Cain provides the solvers HomogeneousDirect2DSearch and HomogeneousFirstReaction, but in principle other solvers could be used too. DoStochKitStochSim() accepts the following arguments:
>>> smod.DoStochKitStochSim(endtime=100,frames=10**4,trajectories=False,
IsTrackPropensities=True,customized_reactions=None,
solver=None,keep_stats=False,keep_histograms=False)
>>> smod.PlotSpeciesTimeSeries()
>>> smod.PlotPropensitiesTimeSeries()
This means that we can save the statistics and histograms information that is created by StochKit, but StochPy does not use this data. StochPy offers both the direct method and the tau-leap method of StochKit. These high-level functions can be used in the same way as DoStochSim():
>>> smod.DoCainStochSim()
>>> smod.DoStochKitStochSim()
StochKit MESSAGE: determining appropriate driver...
running $STOCHKIT_HOME/bin/ ssa_direct_small...
StochKit MESSAGE: created output directory
"/home/user/Stochpy/temp/out/ImmigrationDeath_stochkit.xml"...
running simulation...finished (simulation time approx. 0.0330269 s)
>>> smod.DoStochKitStochSim(solver="tau_leaping")
StochKit MESSAGE: determining appropriate driver...
running $STOCHKIT_HOME/bin/tau_leaping_exp_adapt...
StochKit MESSAGE: created output directory
"/home/user/Stochpy/temp/out/ImmigrationDeath_stochkit.xml"...
running simulation...finished (simulation time approx. 0.02966 s)
done!
After a simulation is performed with the Cain or StochKit solvers, StochPy offers almost all data analysis functionalities. Remember that the data is fixed-interval so information about the time between events is completely lost. Both simulators, however, lack full support of events, assignments, SBML format, and reactions more complicated than mass-action kinetics. StochPy returns warnings and errors if problems arise with these simulators:
>>> smod.Model("CellDivision.psc")
>>> smod.DoStochKitStochSim(endtime=3,frames=29019,trajectories=1)
AssertionError: Reaction R6 is not recognized as mass-action,
so add this reaction to the customized_reactions (list) flag
>>> smod.DoStochKitStochSim(endtime=3,frames=29019,customized_reactions=["R6"])
StochKit MESSAGE: determining appropriate driver...
running "c:\model\StochKit2.0.4_WINDOWS\bin\ssa_direct_mixed_small"...
StochKit MESSAGE: compiling generated code...
this will take a few moments...
StochKit MESSAGE: created output directory
"c:\Stochpy\stochkit_out\out\CellDivision_stochkit.xml"..
running simulation...finished (simulation time approx. 0.623178 seconds)
done!
In this example, compilation time of reactions that are not described by mass-action kinetics costs so much time that StochPy is almost five times faster:
>>> smod.DoStochSim(end=3,mode="time",trajectories=1) # Sim. time: 1.77 s
Stochastic Test suite¶
The stochastic test suite from Evans et al. 2008 (The SBML discrete stochastic models test suite) was used to test the algorithms implemented in StochPy. This test suite tests stochastic simulation software on the following points:
- local and global parameters (parameter overloading)
- boundary conditions
- cell compartment volume
- hasOnlySubstanceUnits flag
- math expression parsing
- compartment volume explicitly including in the rate laws
- assignment rules
- time events
- species population events
All 36 tests were successfully passed by StochPy, except test 3.4 where molecule copy numbers are reset whenever the copy number threshold is reached. A similar observation was done by Erhard et al. (2008). StochPy generates also different output for test 1.11. The reason is that StochPy converts species concentrations (HasOnlySubstanceUnits = True) to species copy numbers; species concentrations are multiplied by the volume of the compartment. Different rate equations are therefore obtained which results in different (but correct) output.
Some examples (1.7, 1.19, 3.3, and 3.4) of the stochastic test suite are shown below. These models test the stochastic simulator on multiple species, assignment rules, time events, and species copy number events respectively. A smaller epsilon value (0.01 rather than 0.03) was necessary to get an accurate prediction with the tau-leap method for 1.5.
>>> smod.Model("dsmts-001-07.xml.psc")
>>> smod.DoStochSim(end=50,mode="time",trajectories=1000)
>>> smod.GetRegularGrid()
>>> smod.PlotAverageSpeciesTimeSeries()
>>> smod.Model("dsmts-001-19.xml.psc")
>>> smod.DoStochSim(end=50,mode="time",trajectories=1000)
>>> smod.GetRegularGrid()
>>> smod.PlotAverageSpeciesTimeSeries()
StochPy can also calculate average autocorrelations. An example is shown for the models 3.3:
>>> smod.Model("dsmts-003-03.xml.psc")
>>> smod.DoStochSim(end=50,mode="time",trajectories=1000)
>>> smod.GetRegularGrid()
>>> smod.PlotAverageSpeciesTimeSeries()
>>> smod.GetSpeciesAutocorrelations()
>>> smod.PlotAverageSpeciesAutocorrelations()


Module 4: Cell Division¶
Cell-to-cell variability arises from the inherent stochasticity of biochemical reactions and of cell growth and division. We developed a stochastic simulation algorithm with cell growth and division which are available in StochPy’s cell division module. While the number of cells increases with 2^n, our algorithm simulates the time evolution of one particular cell, i.e. we track one cell lineage over time. The simulated lineage must reflect a well-defined sample of cells in order to relate the statistical properties of this particular lineage to the statistical properties of the whole three or a well defined sample of cells.
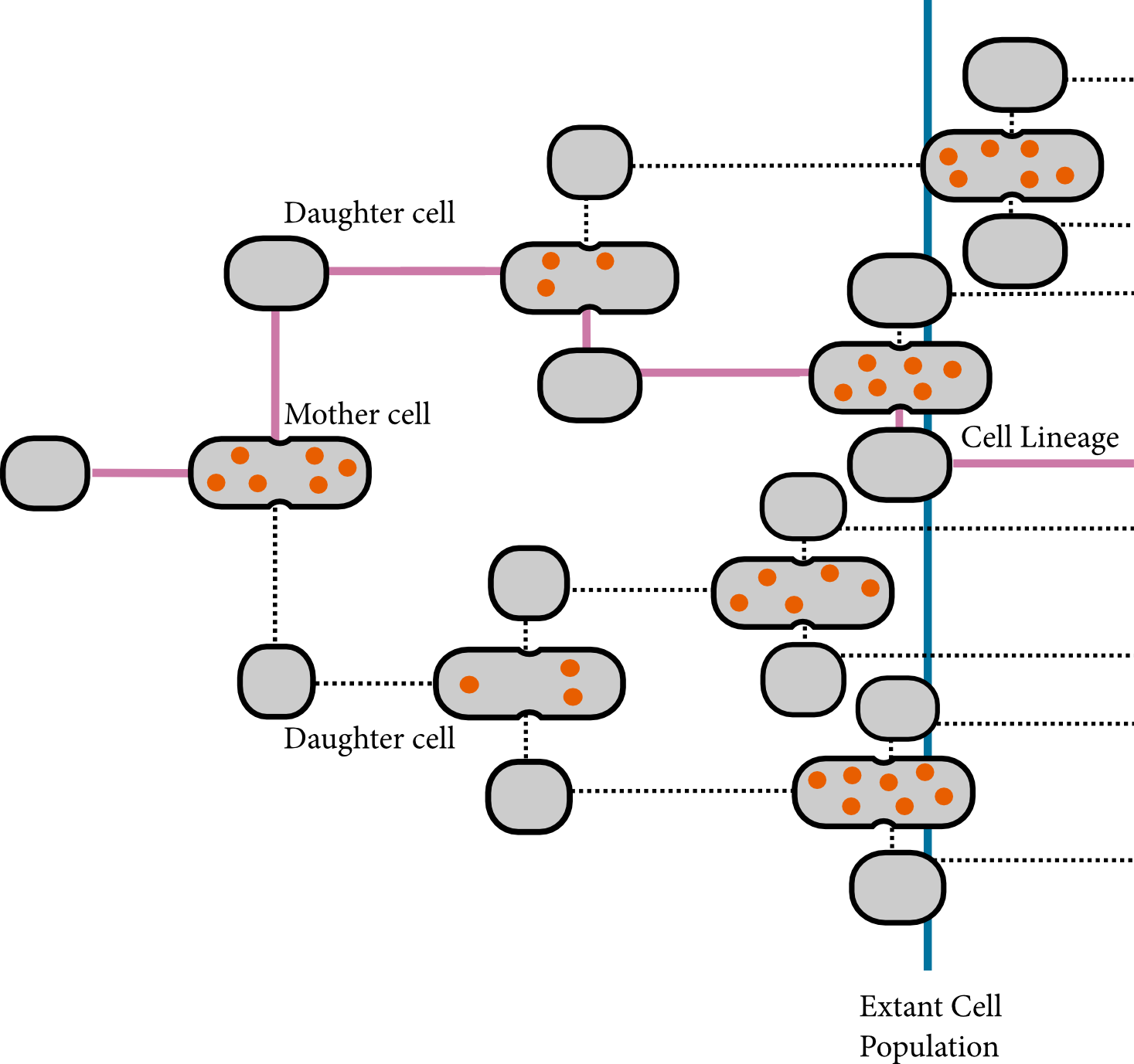
Three different types of samples can be distinguished: samples of extant, baby and mother cells (see also Painter and Marr, 1968). Extant cells are all cells that exist at a specific moment in time, a sample of baby cells consists of all cells that are born within a defined time interval, and sample of mother cells consists of all cells that divide within a defined time interval. In our simulation, we simulate a lineage that represents a sample of mother cells.
StochPy’s cell division module can be started with the following command:
>>> cmod = stochpy.CellDivision()
By default, StochPy uses the model “CellDivision.psc” which describes protein synthesis inside the cell. The StochPy cell division module continues simulating one cell lineage until the number of specified generations, time steps or end time is reached. Modeling of cell division is an example of sequential simulations where each generation starts with the output of the previous generation. This makes these sequential simulations more tricky than doing parallel simulations which is generally easy for most stochastic simulator software packages.
Specifying growth and division properties¶
Doing a stochastic simulation with cell growth and division requires setting, amongst others, growth and division properties. An overview of these settings is shown in the table below. Without specifying these settings, StochPy exploits the default settings shown in the last column.
| Description | StochPy Function | Mandatory Arguments (defaults) |
|---|---|---|
| Growth Function | SetGrowthFunction(growth_rate,growth_type) | (1.0, “exponential”) |
| Initial Cell Volume | SetInitialVolume(initial_volume) | 1.0 |
| Exact Dividing Species | SetExactDividingSpecies(species) | [] |
| Non Dividing Species | SetNonDividingSpecies(species) | [] |
| Set Volume Dependencies | SetVolumeDependencies(IsVolumeDependent, VolumeDependencies,SpeciesExtracellular) | True, [],[] |
| Division properties | SetVolumeDistributions(Phi,K) | (“beta”,5,5), (“beta”,2,2),2) |
Most functionalities are relatively straightforward to use, but we discuss each of them here extensively.
- SetGrowthFunction() sets the specific volume growth rate and the type of growth (exponential or linear).
- SetInitialVolume() sets the initial cell volume (float).
- SetExactDividingSpecies() sets exact dividing species. An example of exact dividing species are chromosomes: In a regular cell division event both daughter cells get one.
- *SetNonDividingSpecies() sets non-dividing species. Specifying non-dividing species can be useful for modeling e.g. DNA.
- SetVolumeDependencies() can be used to override volume dependencies (StochPy determines the order of each reaction automatically). Additionally, this function can be used to set extracellular species. That is, species that are not volume dependent at all and for which no concentrations can be calculated. By default, all species are assumed to be intracellular (and therefore volume dependent if they are second or higher-order reactions).
- SetVolumeDistributions() sets division properties of the cell division volumes and the partitioning distribution between both daughter cells. Phi corresponds to the cell volume distribution at division for a sample of mother cells and K to the partition distribution (Vdaughter1 = Vmother*K and Vdaughter2 = Vmother - Vdaugther1).
The behavior of Phi and K arguments used in SetVolumeDistributions() depends on what type of distribution is provided.
- In the case where both distributions are not beta distributions (all probability distributions from NumPy are available):
- Phi = (“normal”,3,0.1): division volume is normally distributed around 3 with a standard deviation of 0.1;
- Phi = (“fixed,2): division volume is fixed at 2; and
- K = (“fixed,0.75): the volume of the first daughter is always 3/4 of the volume at dvivision and the volume of the second daughter is always 1/4 of the volume at division.
This means that, for instance, Phi = (“fixed”, 2) and K = (“fixed”,0.5) yields a cell lineage where each cells grows from a volume of 1 to 2 (assuming that we also have a initial cell volume of 1). For both distributions, we use by default a beta distribution which is defined on the interval [0,1]. This beta-distribution is symmetrical around 0.5 if both shape parameters are identical.
- In the case where Phi or K are beta distributions:
- Phi = (“beta”,1,1): uniform distribution around the specified mean;
- Phi = (“beta”,2,2): relatively broad distribution around the specified mean; and
- K = (“beta”,100,100): relatively narrow partitioning distribution.
- If both Phi and K are beta distributions, SetVolumeDistributions() requires a third argument that represents the mean of the division volume of the mother cell.
- Phi_beta_mean = 2: a desired volume at division of 2; and
- Phi_beta_mean = 5: a desired volume at division of 5.
We now illustrate how we use these beta distributions:
>>> SetVolumeDistributions(Phi=("beta",5,5),K=("beta",2,2),Phi_beta_mean=2)
In this example, we set a broad distribution for both Phi and K, and specified a mean of 2. The latter means that the cell division event is triggered on average when the cell volume equals 2. We first calculate the Phi_shift: Phi_shift = Phi_beta_mean - 0.5 = 2 -0.5 = 1.5. Next, we can determine the distributions for the mother volume at cell division
and the partitioning distribution
From this, we can calculate the daughter volume just after the cell division event.
In summary, the cell divides at a volume between [1.5,2.5] and the volume at birth is between [0.6,1.5] for these settings.
Run a SSA with cell growth and division¶
After specifying the (default) settings, we can start a stochastic simulation algorithm with explicit cell division in. An overview of this approach is shown in the following figure.

This simulation starts with drawing a next division volume from the specified volume distribution for a sample of mother cells. StochPy subsequently calculates the interdivision time (T) which follows from the chosen volume growth rate, type of growth function, the volume at birth and the next division volume. Assuming that we run a stochastic simulation for a number of generations, we next run a stochastic simulation with cell growth until the cell division event. During the stochastic simulation, the cell volume increases which can have an effect on the likelihood of reactions to fire. When the cell volume at division (drawn from Phi) is reached, StochPy starts with partitioning the cell volume into two daughters. In addition, each species is binomially distributed (weighted by daughter cell volume) between both daughter cells. Then, a next division volume is drawn from Psi and one of the daughters is selected for the next generation. Here, the largest daughter cell is more likely to be selected, because this cell reaches the next division volume faster and is therefore likely to have more descendants. This process continues until StochPy reaches the specified number of generations, time steps or end time. An overview of method is shown above.
From a single lineage simulation to extant-cell population distributions¶
To generate a lineage that is representative for a sample of mother cells, at each division the daughter to be followed by the simulation is chosen with a probability according to the fraction of descendants it can be expected to contribute to the population. Additionally, the following two conditions should be satisfied:
- The first condition requires that cell volume distributions at division and birth are independent. Dependency occurs when a volume at birth is larger than a volume at division, and thus not all volumes from volume distribution at division can be chosen. Independence can be achieved by choosing Phi and K such that there is no overlap possible between cell volumes at division and birth. The simplest way to do this is by choosing a beta distribution for both Phi and K, because this distribution is bounded between 0 and 1 (in contrast to a normal distribution, which is has no bounds).
- The growth law for a single cell is deterministic and independent of concentrations. This is guaranteed by the algorithm.
After simulating the single lineage that is representative for a sample of mother cells, statistical properties of other defined samples (extant, baby) can be calculated with the known relationships of interdivision times and cell age between these samples (more details can be found in our manuscript). In the next section, we provide three examples. In the second and third example, we illustrate analyzing statistical properties for especially the extant cell distribution.
Examples¶
Example 1
We demonstrate the incorporation of cell division for different example models. The first example considers the model “CellDivision.psc” with all default specifications (growth rate, initial cell volume etc.).:
>>> cmod = stochpy.CellDivision()
>>> cmod.DoCellDivisionStochSim()
This model consists of four species, TF, TFactive, mRNA, and Protein. In addition to the existing high-level functions of the stochastic simulation module, several new functionalities are provided within the cell division module. An example is plotting of both species and volume time series. StochPy allows plotting molecule copy numbers and concentrations:
>>> cmod.PlotSpeciesVolumeTimeSeries()
>>> cmod.PlotSpeciesVolumeTimeSeries("concentrations")
The molecule copy numbers plot nicely illustrates illustrates the effect of cell division where each species is binomially distributed between two daughter cells.

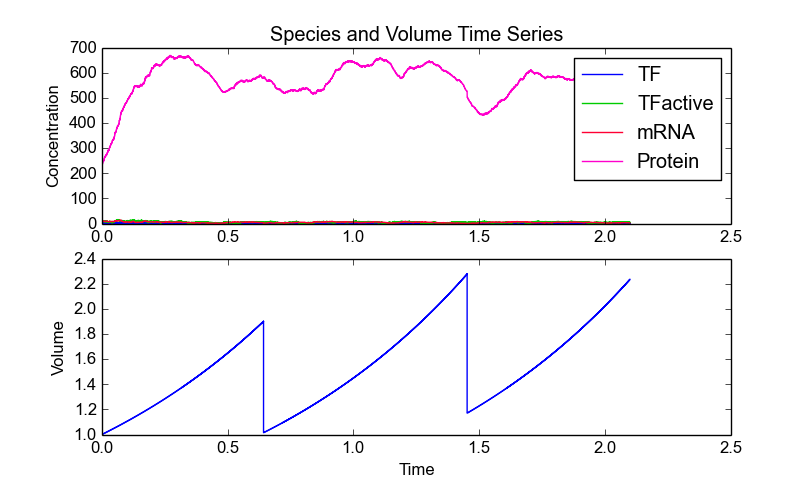
In the volume time series plot, we show the cell volume you expect based on a deterministic growth rate. Importantly, in the stochastic simulation cell volume is only updated if a reaction fires. This means that we underestimate the time until a volume-dependent reaction fires. The reason is that we calculate the volume-dependent propensities with a volume smaller than the volume at the time of the reaction. If we correct for additional volume growth, we would get on average an increased time until firing for these volume-dependent reactions. However, this error is typically very small, because most models have at least one reaction that fires frequently. The error can even be zero if there are no second or higher-order reactions (zero and first order reactions are volume independent).
We can do any type of analysis that is also possible in the SSA module.
Example 2
Now, we use the “ImmigrationDeath.psc” model to illustrate some more functionalities of the cell division module. First, we select the model and modify it interactively:
>>> cmod = stochpy.CellDivision()
>>> cmod.Model("ImmigrationDeath.psc")
>>> cmod.ChangeInitialSpeciesCopyNumber("mRNA",11)
>>> cmod.ChangeParameter("Ksyn",2)
>>> cmod.ChangeParameter("Kdeg",0.1)
Second, we set volume and growth statistics:
>>> cmod.SetVolumeDistributions(Phi=("beta",5,5), K=("fixed",0.5),
Phi_beta_mean=2)
>>> cmod.SetGrowthFunction(growth_rate=0.2, growth_type="exponential")
And thirdly, we perform a stochastic simulation with growth and cell division:
>>> cmod.DoCellDivisionStochSim(end=4000, mode="generations")
We generated 4000 generations to get relatively accurate statistical properties (we recommend using 10000 generations). The cell division module offers the analysis of both species and volume statistics. Here, we start with volume statistics:
>>> cmod.PlotVolumeOverview()
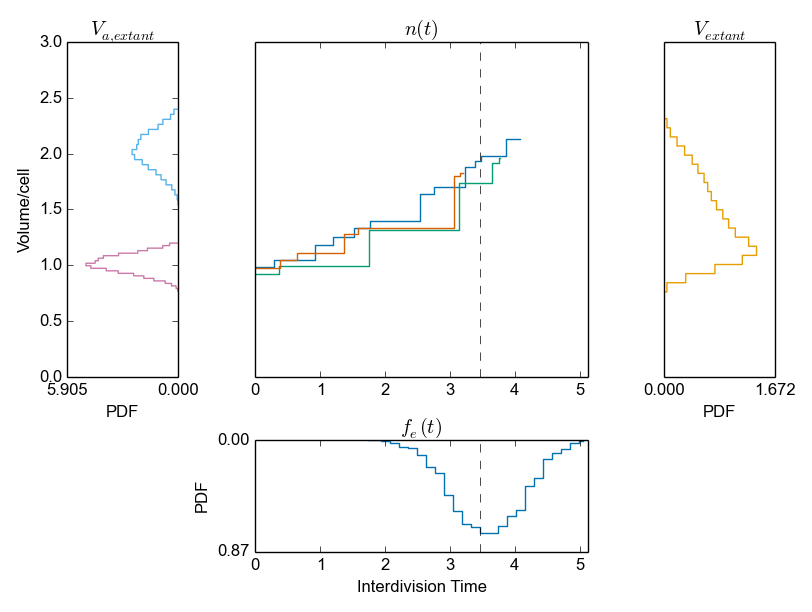
Here, we plot the volume distributions at birth and division for a sample of extant cells (left top panel), the volume time series (center top panel), the volume distribution for a sample of extant cells (right top panel), and the interdivision times for a sample of extant cells (center bottom panel).
Binning and numerical integration is used to relate the sample of mother cells to different samples. StochPy prints a warning if the numerical integration was not accurate enough. Using AnalyzeExtantCells() with a different number of bins for both age and interdivision times should solve this issue.
>>> cmod.AnalyzeExtantCells(n_bins_age=20,n_bins_IDT=20,n_bins_volume=20)
By specifying a different sample, the same plot is generated whereas for a different sample:
>>> cmod.PlotVolumeOverview(sample="mother")
>>> cmod.PlotVolumeOverview(sample="baby")
Plotting functions also exist to plot each of these panels individually: PlotVolumeAtBirthDistribution() , PlotVolumeAtDivisionDistribution(), PlotVolumeTimeSeries(), PlotVolumeDistribution(), and PlotInterdivisionTimeDistribution().
For species, a similar high-level function exists:
>>> cmod.PlotSpeciesOverview()
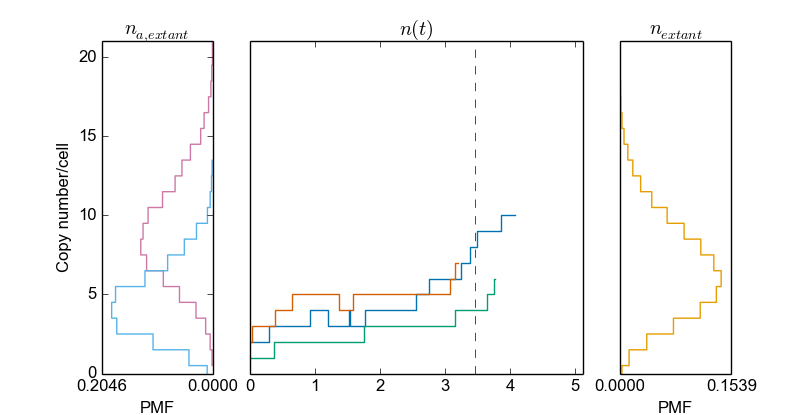
Here, we plot the species distribution at birth and division for a sample of extant cells (left panel), the species time series (center panel), and the species distribution for a sample of extant cells (right panel). Again, by specifying a different sample, the same plot is generated whereas for a different sample:
>>> cmod.PlotSpeciesOverview(sample="mother")
>>> cmod.PlotSpeciesOverview(sample="baby")
Also, plotting function exist to plot each of these panels individually: PlotSpeciesAtBirthDistributions() , PlotSpeciesAtDivisionDistributions(), PlotSpeciesTimeSeries(), PlotSpeciesDistributions(). Note that volume plots are singular and species plot plural; there is only one cell volume and there can be different molecular species inside the cell.
>>> cmod.PlotCellAgeDistribution()

We can also calculate the mean and standard deviation of each species for samples of extant and mother cells. Again, StochPy returns by default the statistics for a sample of extant cells:
>>> cmod.PrintSpeciesMeans()
mRNA 6.86
>>> cmod.PrintSpeciesStandardDeviations()
mRNA 2.82
>>> cmod.PrintSpeciesMeans(sample="mother")
mRNA 6.93
In this particular case, they are slightly different, i.e. the mean of the cell lineage is slightly greater.
Example 3
In the third and final example of the cell division module, we illustrate the effect gene duplications. We use an extended immigration-death model which consists of two different mRNAs that are both transcribed from a different part of the genome. Gene duplication doubles the transcription rate of the corresponding mRNA. We add gene duplication for mRNA1 and mRNA2 which occur as 25% and 75% of the interdivision time, respectively. We expect that in steady state that mRNA1 > mRNA2.
>>> cmod = stochpy.CellDivision()
>>> cmod.Model("GeneDuplication.psc")
>>> cmod.SetGrowthFunction(0.2)
>>> cmod.SetGeneDuplications(["G1","G2"],[0.25,0.75]
>>> cmod.DoCellDivisionStochSim(end=3,method="direct")
>>> cmod.PlotSpeciesTimeSeries()

The time series plot confirms that G1 and G2 are duplicated after 25% and 75% of the interdivision time, respectively. Next, we perform a simulation for 4000 generations to illustrate the differences in mRNA1 and MRNA2 copy numbers:
>>> cmod.DoCellDivisionStochSim(end=4000,mode="generations")
>>> cmod.PrintSpeciesMeans()
G1 1.757
mRNA1 44.849
G2 1.294
mRNA2 31.762
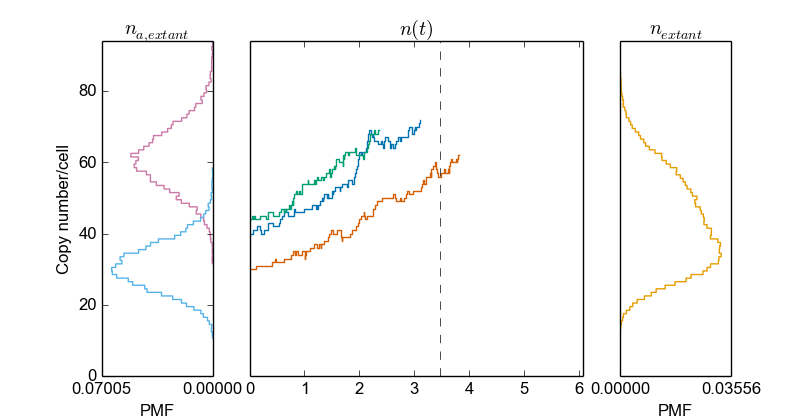
>>> cmod.PlotSpeciesOverview("mRNA1")
>>> cmod.PlotSpeciesOverview("mRNA2")

Module 5: SBML2PSC¶
Models are often described in the SBML format, which makes it difficult to interpret. StochPy offers the SBML2PSC module that can used to convert models written in SBML to models written in the human interpretable PySCeS MDL:
>>> sbml2psc_mod = stochpy.SBML2PSC()
>>> sbml2psc_mod.SBML2PSC(sbmlfile="dsmts-003-03.xml",
sbmldir=stochpy.model_dir, pscfile=None, pscdir=None)
csymbol time defined as "t" in event
Info: single compartment model: locating "Dimerisation" in def. compartment
Info: single compartment model: locating "Disassociation" in def. compartment
Writing file: /home/user/Stochpy/pscmodels/dsmts-003-03.xml.psc
SBML2PSC
in : /home/user/Stochpy/pscmodels/dsmts-003-03.xml
out: /home/user/Stochpy/pscmodels/dsmts-003-03.xml.psc
The PySCeS MDL (see the PySCeS Model Description Language section) is the default model description format, but StochPy can parse SBML models.
Modeling Input¶
Models¶
In the previous sections, we used existing models to illustrate how StochPy can be used for stochastic modeling. Here, we briefly discuss what kind of models StochPy accepts. Deterministic rate equations have normally been used to describe a system of biochemical reactions, whereas these equations are often not valid for stochastic modeling. We advice potential users of StochPy that are unaware of the differences between deterministic and stochastic rate equations to read section Stochastic Rate Equations carefully.
In short, for stochastic simulators the input consists of irreversible rate equations and initial copy numbers. StochPy automatically converts concentrations into molecule copy numbers. We advice users of StochPy to do not use net stoichiometric coefficients in the model description. While the StochPy solvers handle them correctly, the fixed-interval solvers that we use of other software packages cannot handle them correctly.
StochPy supports models written in both PySCeS MDL and SBML and therefore also SBML event facilities (see here) and assignments (see here). The PySCeS MDL is used as default format which is further explained in the PySCeS Model Description Language section. Not all functionalities are supported by StochPy (e.g. piecewise functions). Correct installation of libSBML is required before StochPy supports parsing of SBML models. More details can be found in the Installation section. Models written in SBML are automatically converted into the PySCeS MDL format which are subsequently used by the stochastic simulators.
StochPy provides many example models, which can be used as a reference for building your own models. We show here two examples:
# Reactions
R1:
$pool > mRNA
Ksyn
R2:
mRNA > $pool
Kdeg*mRNA
# Variable species
mRNA = 50.0
# Parameters
Ksyn = 10
Kdeg = 0.2
We call this the Immigration-Death model which we use as the default model in StochPy. First, we define two reactions: a zero-order reaction R1 synthesizing the species mRNA and a first-order reaction R1 degrading mRNA. Second, we set an initial mRNA copy number. And third, we set the parameter values of the synthesis and degradation rate. The next example is the Birth-Death model:
# Compartments
Compartment: Cell, 1.0, 3
# Reactions
Death@Cell:
X > $pool
Mu*X
Birth@Cell:
X > {2} X
Lambda*X
# Fixed species
# Variable species
X@Cell = 100.0
# Parameters
Mu = 0.11
Lambda = 0.1
We first define a compartment called “Cell” where all reactions take place. Now, both reactions are first order reactions. The birth reaction is an example where we can use a net stoichiometry ($pool > X).
Events¶
StochPy supports both time and species copy number events (for more details see here). Here, we illustrate these events with a model of dimerization of P to P2. The initial numbers of molecules of P and P2 are 100 and 0 respectively. We use model 3.3 from the dsmts test suite, but to illustrate the difference with and without (time) event we first remove the time event:
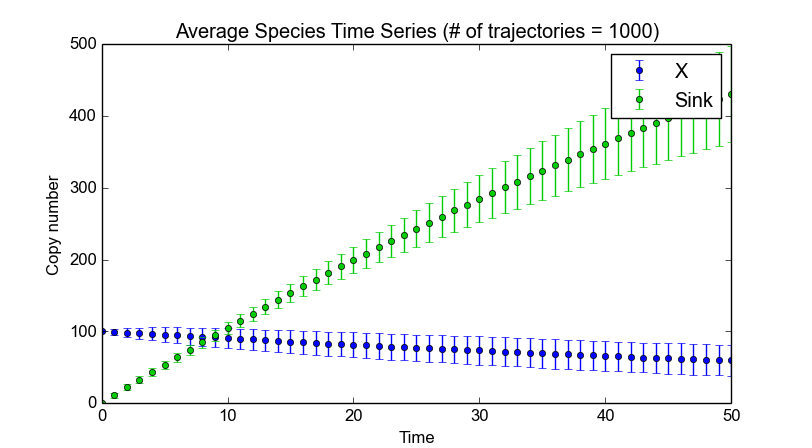
>>> smod.Model("dsmts-003-03.xml.psc")
>>> smod.DoStochSim(end=50,mode="time",trajectories=1000)
>>> smod.GetRegularGrid()
>>> smod.PlotAverageSpeciesTimeSeries()

The figure above shows the outcome of our simulations. We now put back the time event in model 3.3 of the dsmts test suite. This time event resets the molecule numbers of P and P2 at t>=25. Events have access to the “current” simulation time using the _TIME_ symbol, which gives the following description of an event in the PySCeS MDL:
# Event definitions
Event: reset1, _TIME_ >= 25, 0.0
{
P2 = 0
P = 100
}
The figure above shows the effect of adding the time event: the copy numbers of both molecular species are reset at t=25.
A second example is a species event where we reset the molecule numbers once molecule P is greater than 30:
# Event definitions
Event: reset2, P2 > 30, 0.0
{
P2 = 0
P = 100
}
This event is incorporated in the model 3.4 from the dsmts test suite. Stochastic simulation with StochPy yields the following result:
>>> smod.Model("dsmts-003-04.xml.psc")
>>> smod.DoStochSim(end=50,mode="time",trajectories=1000)
>>> smod.GetRegularGrid()
>>> smod.PlotAverageSpeciesTimeSeries()

So far, we used a time or species trigger but we can also combine both type of events:
# Event definitions
Event: reset3, P2 > 30 and _TIME_ > 20, 0.0
{
P2 = 0
P = 100
}
Species are now reset if P2 > 30 and if t>20. This event can also occur multiple times.
Warning: do not use or specifications in event triggers, but use separate events instead.
StochPy also allows for the mutation of parameters. Define this parameter as a “fixed species” (StochPy gives a warning that this “fixed species” does not occur in any reaction). This warning message is correct (it’s a parameter, not a species). Next, you can add a time event to modify the parameter during the simulation:
Event: jump1, _TIME_ >= 25, 0.0
{
k1 = 0.01
}
In this example, k1 is set to 0.01 at t=25 without any delay.
Assignments¶
Now, we shortly describe one example of an assignment rule for which we use a simple birth-death process of metabolite X. The assignment rules defines that a new species y = 2 * X:
# Assignment rules
!F y = 2.0*X
This assignment is incorporated in the dsmts-001-19.xml.psc file and simulation yields the following result.
Stochastic Rate Equations¶
In this section, we compare deterministic and stochastic rate equations. In short, they are identical for zero and first-order reactions, but not for second and higher-order reactions (if a two or more of the same species are necessary for the reaction to take place, e.g. 2X > Y).
Deterministic models represent species often by concentration, while stochastic models represent species often by molecule copy numbers. The molecule copy numbers are identical to:
Na*[X]*Volume(L)
Here, Na is the number of Avegadro (6.02*10e23). This is the first step of the conversion of deterministic rate constants (k) to stochastic rate constants (c).
Zero-order reaction¶
Assume the following reaction:
--> X
The deterministic rate equation of this reaction is:
k (Ms^-1)
The stochastic rate equation of this reaction is:
c (s^-1)
First-order reaction¶
Consider the following reaction:
X --> Y
The deterministic rate equation of this reaction is:
k*[X]
The stochastic rate equation of this reaction is:
c*x (s^-1)
Here, x corresponds to Na*[X]*Volume(L) particles. Therefore, x changes k*Na*[X]*Volume(L) = k*x molecules per second, which means that k=c. This means that first order stochastic and deterministic rate equations are identical.
Second-order reaction¶
There exist several types of second-order reactions. First, consider the next reaction:
X+Y --> Z
The deterministic rate equation of this reaction is:
k*[X]*[Y] (Ms^-1)
The stochastic rate equation of this reaction is:
c*x*y (s^-1)
Again, [X] corresponds to Na*[X]*Volume(L) particles and the same is true for [Y]. Therefore, the following relationship holds:
k*[X]*[Y] (Ms^-1) = (k*x*y)/(Na*V)^2, hence c = k/(Na*V)
Secondly, consider a dimerization reaction:
2X --> Y
The deterministic rate equation of this reaction is:
k*X^2
The stochastic rate equation of this reaction is:
0.5*c*x*(x-1)
The first important concept to understand is that a dimerization reaction can only occur if there are at least two molecules of species X available. For this reason, the stochastic rate equation must be zero if there is only one X molecule available.
Third-order reaction¶
This type of reactions does usually not occur in chemical reactions, but they are described just for the sake of completeness.
There exist three types of third-order reactions
1) X+Y+Z –> A
The deterministic rate equation of this reaction is:
k*[X]*[Y]*[Z]
The stochastic rate equation of this reaction is:
c*x*y*z
2) X+2Y –> Z
The deterministic rate equation of this reaction is:
k*[X]*([Y]^2)
The stochastic rate equation of this reaction is:
0.5*c*x*y*(y-1)
3) 3X –> Y
The deterministic rate equation of this reaction is:
k*[X]^3
The stochastic rate equation of this reaction is:
(1/6)*x*(x-1)*(x-2)
For the last reaction three x molecules are necessary, thus this rate equation can not fire if there are less than three molecules of x available.

StochPy User guide
Navigation
- Introduction
- Installation and Configuration
- Using StochPy
- Modeling Input
- The PySCeS Model Description Language